


Work with us
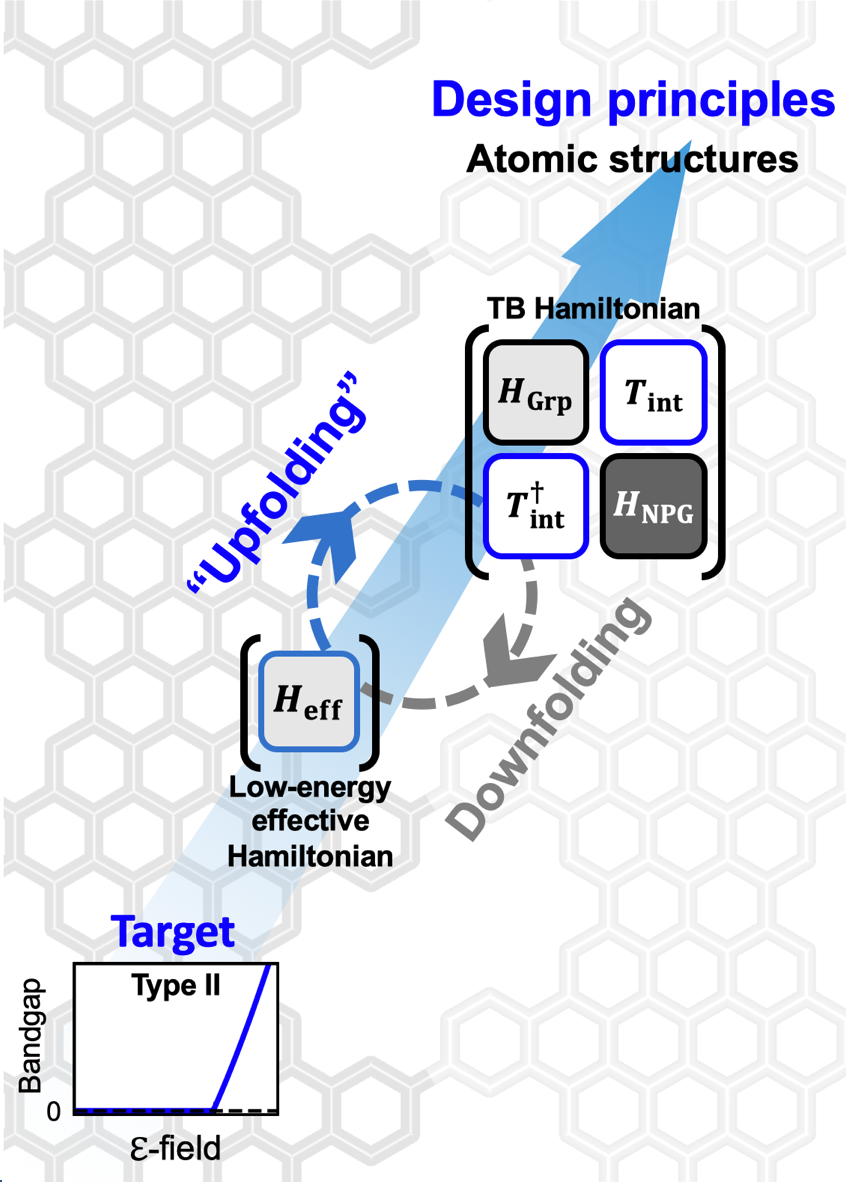
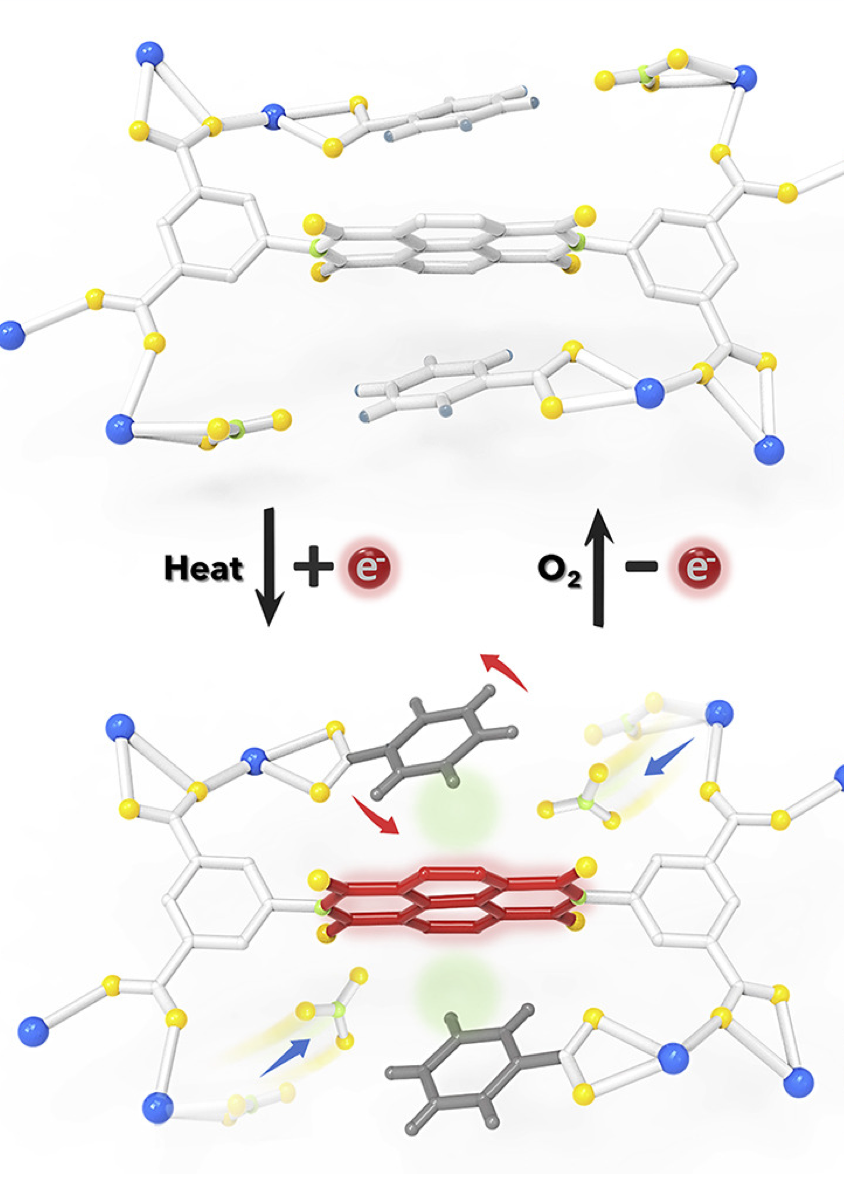
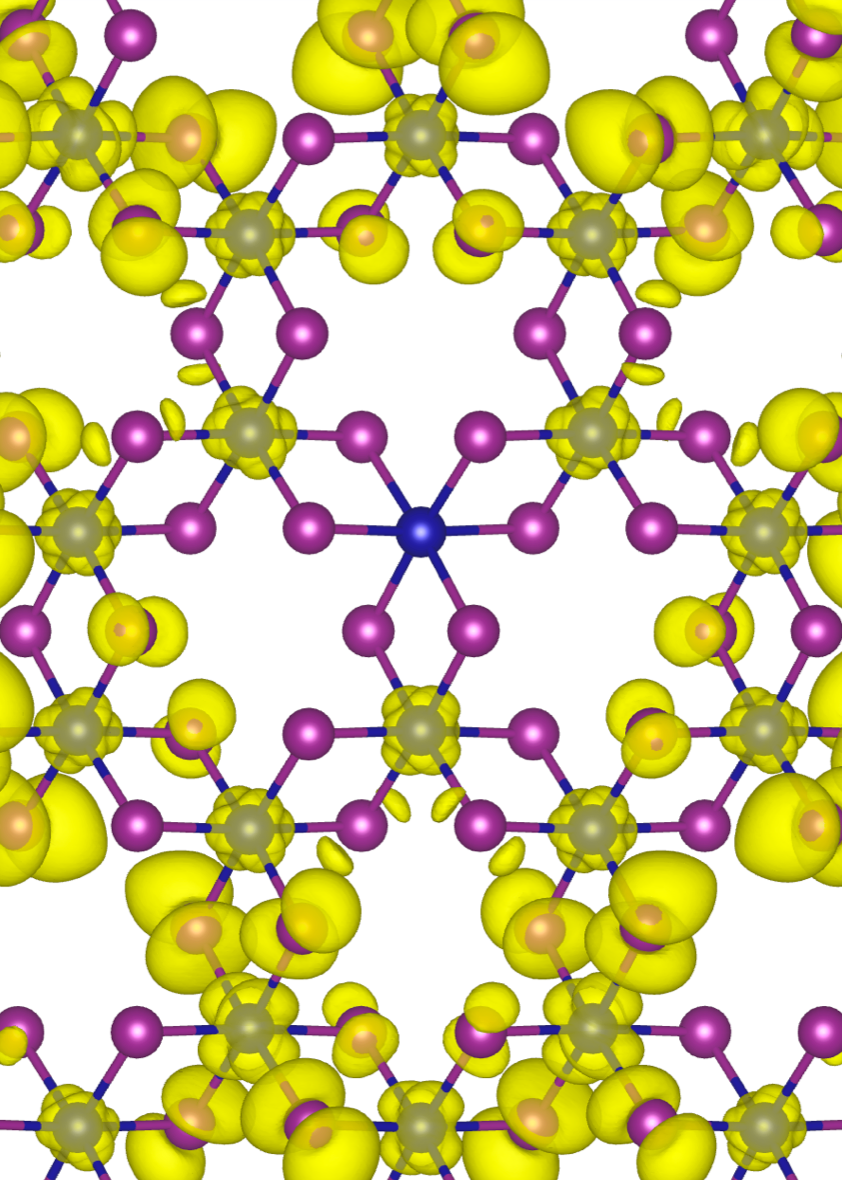
Our research intersects materials physics, chemistry, and machine learning. We employ quantum and statistical mechanics, alongside machine learning, to understand and predict materials properties.
All graduate students at DGIST are eligible for fully funded graduate scholarships. Admission information can be found here (in English) or here (in Korean). We invite prospective students to get in touch with Prof. Kang at joongoo.kang@dgist.ac.kr. To learn more about our research, you may refer to Publication List.
All graduate students at DGIST are eligible for fully funded graduate scholarships. Admission information can be found here (in English) or here (in Korean). We invite prospective students to get in touch with Prof. Kang at joongoo.kang@dgist.ac.kr. To learn more about our research, you may refer to Publication List.